Introduction
Decades of research have shown that dimethylsulfide (DMS) in the atmosphere originates with phytoplankton in the ocean. Once in the atmosphere, DMS is oxidized to particulate non-sea salt (nss) sulfate (SO42–), which affects climate by scattering incoming solar radiation and nucleating cloud droplets. The NOAA Pacific Marine Environmental Laboratory (PMEL) has organized and participated in research cruises for four decades to characterize seawater DMS concentrations and emissions as well as aerosol sources, properties, atmospheric processing, and climate impacts in the atmospheric marine boundary layer (MBL). Figure 1 shows a map of relevant cruise tracks that cover all ocean basins. This paper highlights PMEL’s findings on the climate roles of nss-SO42– and sea spray aerosol in the MBL and summarizes the influence of PMEL’s measurements on what is known today about marine atmospheric aerosol particles. Future directions are also discussed.
Throughout the paper, the term “marine aerosol” refers to all aerosol chemical components observed in the marine atmosphere regardless of origin (natural, anthropogenic, or continental).
Seawater DMS
Emission of Oceanic DMS Is the Largest Natural Source of Sulfur to the Atmosphere
In 1979, PMEL conducted a cruise in Cook Inlet, Alaska, to assess background concentrations of benzene and toluene in the water column before the start of oil and gas leases in the region (Katz and Cline, 1981). Using a new gas chromatograph–mass spectrometer (GCMS), a mysterious peak overwhelmed the target compounds. This compound was determined to be DMS, although these early results were not published. The only published measurements of seawater DMS at that time were reported by Lovelock et al. (1972) from sites along the southern coast of England and across the Atlantic between South America and the United Kingdom. Motivated by the Alaska findings, PMEL developed a purge and trap system for collecting seawater DMS, with chemical analysis by a flame photometric detector gas chromatograph (FPD GC) to specifically quantify sulfur compounds in the surface ocean (Cline and Bates, 1983).
Between 1982 and 1985, cruises were conducted in the North Pacific, Gulf of Alaska, and South Pacific to assess seasonal and latitudinal distributions of concentrations of DMS in the surface ocean and to calculate its flux to the atmosphere (Bates et al., 1987). These data were used to estimate the first ocean sulfur fluxes to the atmosphere, taking into account area-weighted, seasonal DMS concentrations. Bates et al. (1992) went on to combine these data with those from additional studies to create a global inventory of sulfur emissions from natural sources, both oceanic and terrestrial. It was found that oceanic DMS was the largest natural source of sulfur to the atmosphere, making up about 15% of the combined natural and anthropogenic sources. In contrast, the terrestrial biogenic source was about 2%. Terrestrial sources include salt- and freshwater swamps, soils, and plants (Yoch, 2002). During these cruises, indicators of primary productivity (e.g., chlorophyll and nutrients) were found to be broadly correlated with DMS, indicating that the production of DMS was linked to phytoplankton abundance.
The Ocean-to-Atmosphere Flux of DMS Is a Minor Sink in the Seawater Sulfur Cycle
PMEL’s next step was to investigate the processes controlling the cycling of sulfur in surface seawater. During two cruises (Northeast and equatorial Pacific), measurements of key sulfur species in surface seawater were made along with rates of conversion between these species (Bates et al., 1994; Kiene and Bates, 1990). These measurements allowed for the most complete picture of the sulfur cycle at the time. It was found that most seawater DMS is microbially consumed in the water column, while the ocean-to-atmosphere flux is a minor sink in the seawater sulfur cycle (Figure 2).
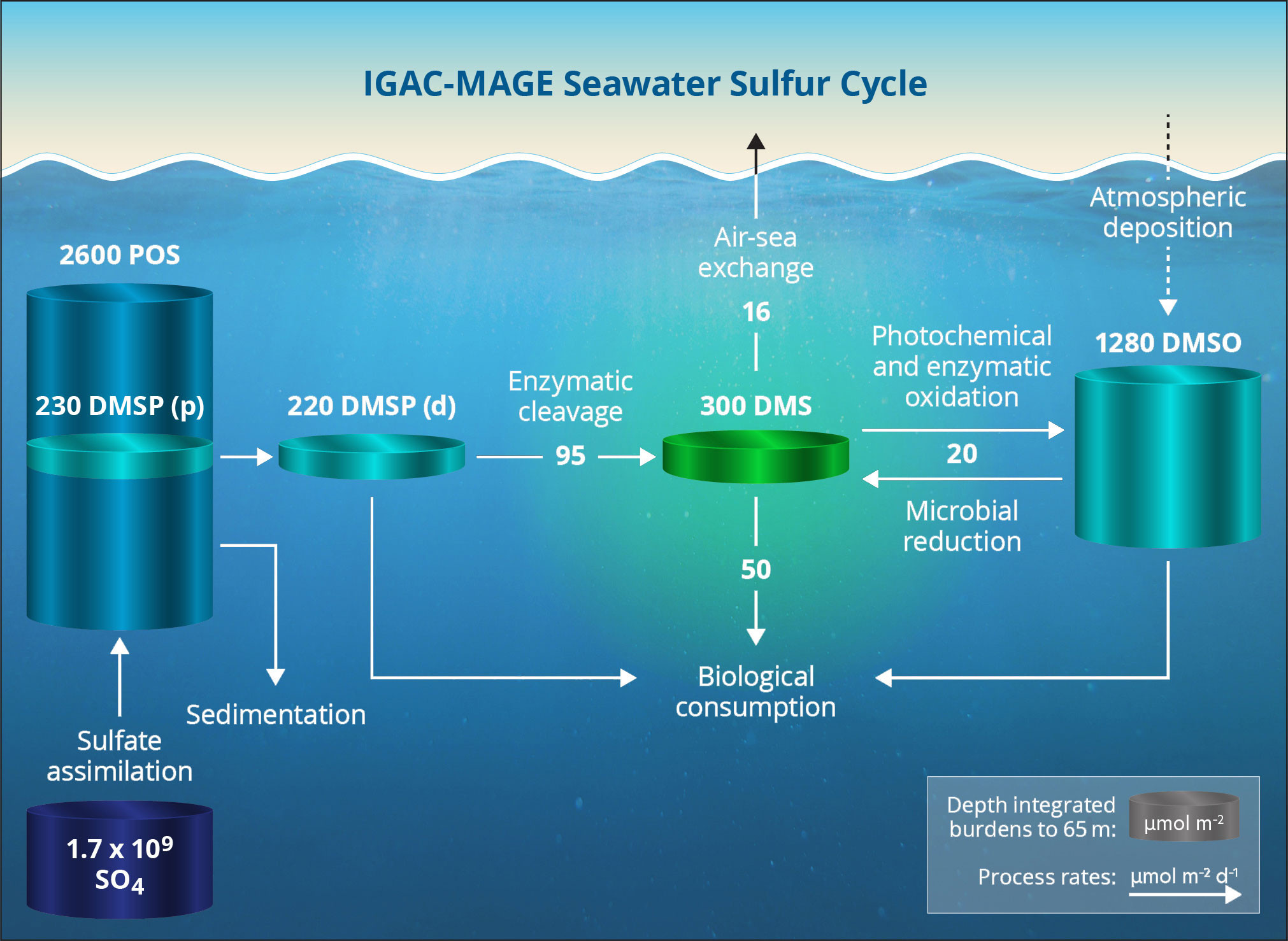
FIGURE 2. Seawater sulfur cycle as determined by Bates et al. (1994). Concentrations are average depth-integrated amount (60 m) in μmol m–2. Processes affecting concentrations are in μmol m–2 d–1. Bacteria and phytoplankton assimilate dissolved sulfate. A portion of the assimilated sulfate is incorporated into dimethylsulfoniopropionate (DMSP). DMSP is enzymatically cleaved to form dimethylsulfide (DMS). DMS is then biologically consumed, emitted to the atmosphere, or converted to dimethylsulfoxide (DMSO). > High res figure
|
Atmospheric Marine Aerosols
PMEL added aerosols to shipboard measurements in the mid-1980s to investigate the fate of DMS once emitted to the atmosphere. At the time, little was known about the sources and composition of aerosol over the open ocean. Early shipboard (Parungo et al., 1986), aircraft (Andreae et al., 1988), and island station (Savoie and Prospero, 1989) measurements over the North and South Pacific Oceans indicated that DMS was the only source of nss-SO42– to the MBL. The role of long-range transport was not widely acknowledged.
Continental Aerosols Undergo Long-Range Transport to the Remote Ocean
In spring of 1988, PMEL made the first open-ocean simultaneous measurements of the main seawater, gas phase, and particulate phase species involved in interactions between the sulfur and reduced nitrogen cycles over a wide latitudinal range (53°N to 14°S along 170°W; Quinn et al., 1990). These measurements were among the first to definitively link long-range transport of anthropogenic sulfate to the central Pacific. Calculated air mass back trajectories (Stein et al., 2015) along the southernmost portion of the cruise track (14°N and 11°S) showed easterly flow, with no passage of the sampled air masses over land for at least four days before reaching the ship. In contrast, along the northernmost portion of the cruise track (50°N to 30°N), mean nss-SO42– concentrations were a factor of three higher. As indicated by air mass trajectories, the higher concentrations were due to long-range transport from Asia to the central Pacific, indicating that submicron aerosols have a sufficient lifetime to be transported thousands of kilometers. Up to the late 1980s, climate models were limited to greenhouse gases (Ramaswamy et al., 2019). Evidence such as elevated concentrations of nss-SO42– over the remote ocean helped prompt the inclusion of aerosols into global models (Charlson et al., 1991).
Large and Variable Fraction of Submicron Marine Aerosol Is Not Sulfate
Initially, most model estimates of direct aerosol radiative forcing only included a highly simplified anthropogenic sulfate-only aerosol (IPCC, 2021). Knowledge of the relative mass concentrations of all chemical components present and their variability improves the accuracy of estimates of aerosol radiative forcing. PMEL used measurements of submicron aerosol gravimetric mass and mass concentrations of inorganic ions from several cruises (RITS93, RITS94, ACE-1, CSP; Figure 1) to determine mass fractions of nss-SO42–, sea salt, and a residual component calculated as the difference between the total gravimetric aerosol mass and the sum of the mass of analyzed components (Quinn et al., 2000). Only submicron aerosol was included because it is the size range most efficient at scattering and absorbing sunlight. It was found that the submicron mass fractions of nss-SO42–, sea salt, and the residual component were similar, ranging between 0.07 and 0.53. It was hypothesized that the bulk of the residual mass consisted of organics, which, depending on hygroscopicity, could impact cloud nucleating abilities and magnitude of light scattering. These results indicated that a large and variable fraction of submicron aerosol in the marine atmosphere is not sulfate, indicating that all species need to be included in estimates of aerosol radiative forcing in the marine atmosphere.
Submicron Sea Salt Is a Major Contributor to Light Scattering in Marine Regions
The wind-driven production of sea spray aerosol results in one of the largest global sources of primary atmospheric aerosol particles on a mass concentration basis (Warneck, 1988). Sea spray is composed of the inorganic salts and organic matter found in seawater (Quinn et al., 2015). Initial global chemical transport and climate models only included sea salt with particle diameters greater than ~2 µm due to “few measurements of sea salt size spectra” (Tegen et al., 1997). Assuming a minimum diameter of 4 µm for sea salt particles, Tegen et al. (1997) found that the contribution of sea salt to light extinction was small (<28%) on global and regional scales. In response, PMEL calculated the contribution of sea salt to aerosol light scattering based on measurements from cruises in the Pacific and Southern Oceans (PSI-1, MAGE92, RITS93, and RITS94; Figure 1; Quinn and Coffman, 1999). Over all regions, the percent of total scattering by submicron nss-SO42– and of sea salt were similar, ranging from 2.5% to 42% while supermicron sea salt contributed 40% to 64%. This analysis showed that (1) light scattering by submicron sea salt and by nss-SO42– occur at about the same magnitude, and (2) the inclusion of submicron sea salt into climate models is essential for accurate estimates of the impacts of natural aerosols on Earth’s radiation budget. The reported sea salt size distributions and optical properties have since been incorporated into chemical transport and radiative transfer models (e.g., Collins et al., 2001; Zacobson, 2001; Jaegle et al., 2011).
Non-Sea Salt Sulfate Comprises the Majority of the Cloud Condensation Nuclei Population in the Marine Boundary Layer
Sea salt dominates aerosol mass and light scattering in the remote marine boundary layer due to its wind-driven, primary production mechanism (Quinn and Coffman, 1999; Jacobson, 2001). Additional species formed through secondary processes (gas-to-particle condensation and nucleation) contribute to the marine particle number population, resulting in a number size distribution that is a mix of primary sea salt, secondary nss-SO42– derived from DMS, secondary ocean-derived organics, and aerosols emitted from continental sources. The number size distribution and chemical composition of aerosol particles determine their ability to act as cloud condensation nuclei (CCN; i.e., they take up water and nucleate to form cloud droplets). Identifying sources of CCN in the MBL is necessary for understanding regional impacts of aerosols on cloud properties such as effects on albedo and lifetime.
To determine the sources of CCN over the remote ocean, PMEL applied a lognormal-mode-fitting procedure to particle number size distributions measured between 70°S and 80°N on research cruises in the Pacific, Southern, Arctic, and Atlantic Oceans (RITS93, RITS94, ACE-1, ICEALOT, WACS-2, NAAMES-1; Figure 1; Quinn et al., 2017). It was found that the marine boundary layer CCN population is composed mainly of nss-SO42– aerosols produced by large-scale meteorological features that result in the entrainment of particles from the free troposphere. The one exception was the high southern latitudes (70°S to 40°S) where sea spray aerosol comprised up to 65% of the CCN at low supersaturations (0.1%). The relative contributions of different sources to nss-SO42– in the MBL remains an open question, with candidates that include oxidation of biogenic or anthropogenic sulfate precursors in the free troposphere with subsequent entrainment into the MBL and/or long-range transport of anthropogenic emissions.
A DMS-Controlled Marine Biota-Climate Feedback Is Unlikely to Exist
The established connection between surface seawater phytoplankton, production of DMS, and the oxidation of DMS to form nss-SO42– aerosol led to a hypothesis put forth in the 1980s that marine biota could be involved in a climate feedback loop. The CLAW hypothesis, named after the four authors of the Charlson et al. (1987) study, noted that marine phytoplankton produce the osmolyte dimethylsulfoniopropionate (DMSP), which undergoes enzymatic cleavage in surface seawater to form DMS (Figure 2). DMS is then emitted to the atmosphere where it is oxidized to form nss-SO42– aerosol so that an increase in DMS emissions from the ocean could result in an increase in CCN, cloud droplet number concentration, and albedo, and a decrease in the amount of solar radiation reaching the ocean surface. It was hypothesized that this reduction of solar radiation could result in a change of phytoplankton speciation and abundance, thus setting up a feedback loop between cloud albedo and surface seawater DMS concentrations.
After years of seawater and atmospheric measurements, it became clear that aerosols in the MBL are much more complex than assumed by the CLAW hypothesis (Quinn and Bates, 2011). There are sources of CCN in the MBL not derived from DMS, including sea spray and anthropogenic aerosol. Charlson et al. (1987) assumed a highly simplified relationship between CCN and cloud albedo. It is now known that aerosols affect not only cloud microphysics and albedo but also cloud macrophysics (cloud fraction, size, and morphology; Zuidema et al., 2008; Small et al., 2009). Neglecting these aerosol impacts on clouds likely led Charlson et al. (1987) to significantly overestimate the sensitivity of cloud albedo to a change in DMS emissions. The CLAW hypothesis also assumed that changes in cloud albedo, surface ocean temperature, or incident solar radiation led to changes in the production of DMS. Several model studies published after Charlson et al. (1987) have shown that surface seawater DMS concentrations have a low sensitivity to changes in all of these factors (Gunson et al., 2006; Vallina et al., 2007). In addition, the formation of new particles via DMS oxidation to nss-SO42– occurs primarily in the free troposphere in cloud outflow regions with subsequent transport and entrainment to the MBL (e.g., Clarke et al., 1998). As a result, a local connection between surface ocean phytoplankton and cloud albedo is not likely.
After 25 years of research by atmospheric scientists and marine biologists, PMEL’s synthesizing article retired the CLAW hypothesis due to a lack of evidence for a DMS-controlled marine biota-climate feedback (Quinn and Bates, 2011). Figure 3 shows an updated view of the multiple sources of CCN to the MBL. The CCN population in the MBL is a result of emissions of sea spray aerosol that includes the inorganic salts and organic matter found in surface seawater, entrainment of biogenic and anthropogenic sulfate from the free troposphere, and long-range transport of other chemical components.
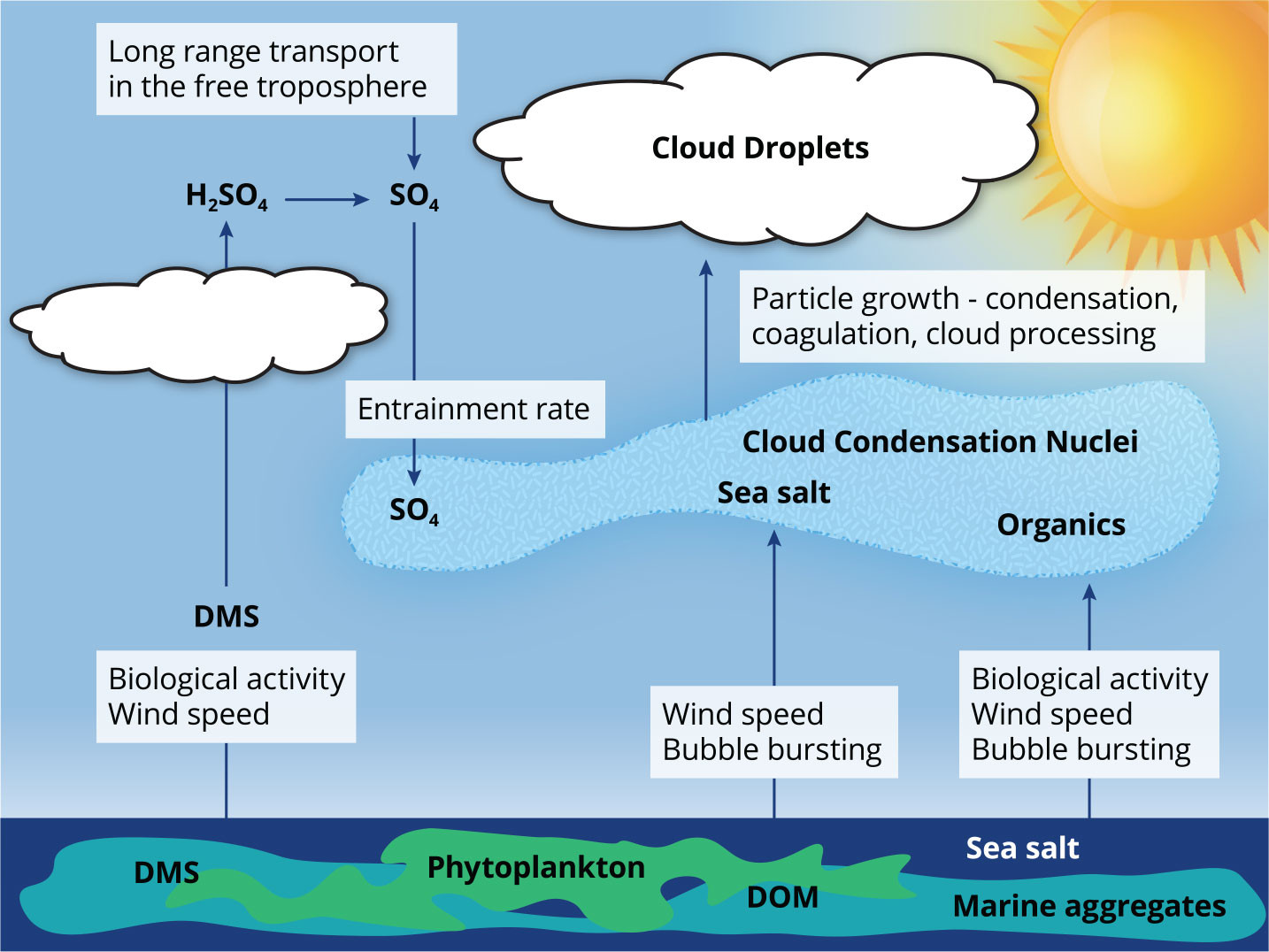
FIGURE 3. Major sources of cloud condensation nuclei (CCN) in the atmospheric remote marine boundary layer. DMS contributes to the boundary layer CCN population primarily via particle nucleation in the free troposphere with subsequent subsidence. Sea salt and organics are emitted as a result of wind-driven bubble bursting at the ocean surface. DMS = Dimethylsulfide. DOM = Dissolved organic matter. > High res figure
|
Marine Ecosystems Have Little Effect on Sea Spray Aerosol Emission Flux, Organic Fraction, and CCN Activity
Quinn and Bates (2011) left open the possibility that sea spray aerosol could provide a link between ocean-derived CCN and climate. The direct emission of sea spray organics into the atmosphere could allow for surface ocean biological production to impact CCN production and cloud albedo. PMEL developed a novel over-the-side method for sampling fresh sea spray aerosol emissions by generating breaking waves in situ alongside a ship (Bates et al., 2012). Sea Sweep, the sea spray aerosol generator, was used on five cruises to sample sea spray aerosol during different stages of the North Atlantic plankton bloom (Bates et al., 2020; Figure 4). The cruises (WACS-2, NAAMES-1 to 4; Figure 1) covered extreme surface ocean conditions from the oligotrophic Sargasso Sea to the biologically productive western subarctic.

FIGURE 4. Deployment of Sea Sweep from Woods Hole Oceanographic Institution’s R/V Atlantis off the coast of California. > High res figure
|
It was found that seasonal differences in North Atlantic plankton ecosystems had very little impact on sea spray aerosol properties, including organic mass fraction, cloud-nucleating ability, and number production flux (Bates et al., 2020). Given the relatively invariant sea spray aerosol organic mass fractions, it was concluded that the ocean source of the organics is the large pool of dissolved organic carbon instead of particulate organic carbon that varies with phytoplankton biomass. As a result, chemical transport and climate models can consider the ocean to provide a uniform source of sea spray aerosol organics, greatly simplifying estimates of radiative effects.
Future Directions
A full understanding of aerosol direct and indirect radiative forcing in the marine atmosphere is not possible without measurements of vertical profiles of aerosol and cloud properties. Over the past decade, PMEL has developed payloads for uncrewed aerial systems (UASs) for the measurement of vertical profiles of aerosol and cloud properties. In tandem, PMEL has worked with the NOAA Uncrewed Aerial Systems Program, UxS Operations Center, Small Business Innovation Research (SBIR) Program, and L3Harris to develop a UAS for shipboard use. Successful, fully autonomous shipboard operation of L3Harris’ hybrid Fixed-Wind-Vertical-Take-Off-and-Landing (FVR-55) with PMEL payloads onboard was demonstrated in March of 2022 offshore of Key West (Figure 5). The first over-the-open-ocean science flights took place from the Tillamook UAS Test Range near the coast of Oregon in August 2022. Future directions include implementing lower atmosphere shipboard measurements with routine vertical profiling.

FIGURE 5. FVR-55 uncrewed aerial system in flight with the Cloudy Sky payload onboard offshore of Key West. > High res figure
|
Conclusions
Four decades of measurements by PMEL have revealed the climate roles of non-sea salt sulfate and sea spray aerosol in the atmospheric MBL. These measurements have led to the largest global ocean database of surface seawater DMS concentrations (https://saga.pmel.noaa.gov/dms/) and aerosol properties (https://saga.pmel.noaa.gov/data/). These data have and will continue to be used extensively by the wider atmospheric sciences community. Going forward, PMEL aspires to add vertical profiles of aerosol and cloud properties to the existing database.
Acknowledgments
We thank the crews of the numerous ships that made this research possible including the NOAA R/Vs McArthur, Discoverer, John Vickers, Surveyor, and Ronald H. Brown, and Woods Hole Oceanographic Institution’s R/V Atlantis. We also thank the many people (D. Covert, D. Hamilton, K. Schulz, V. Kapustin, C. Zenker, J. Benson, M. Hamilton) and funding agencies (NOAA, NASA, and NSF) who contributed to these efforts. This is PMEL contribution number 5472.


