What Information Can Be Gained from Sedimentary Ancient DNA?
Analysis of marine sedimentary ancient DNA (sedaDNA) allows identification of deceased organisms that have sunk from the upper water layers to the bottom of the ocean and become preserved. As a result of the sedimentation process, the remains of deceased organisms accumulate over time, forming a continuous record of past communities that have inhabited the ocean. Marine sedaDNA can be used to study a broad variety of taxa, including viruses, archaea, prokaryotes (bacteria), and eukaryotes (phytoplankton to larger predators). Eukaryotic planktonic organisms, such as diatoms, dinoflagellates, coccolithophores, and foraminifers, are particularly interesting targets for sedaDNA studies because of their established reliability as environmental indicators. However, the breadth of taxa for which genetic signals are preserved in the sediments—including species that do not fossilize—means that sedaDNA holds enormous potential to go beyond these standard environmental proxies and facilitate reconstruction of past marine ecosystems across the food web. Further, in cases where the preservation, or information content, of microfossil, physical, (biogeo)chemical or biomarker proxies is limited, sedaDNA can provide novel insights into past oceanographic and environmental conditions.
What Makes Ancient DNA Different from Modern DNA?
Ancient DNA is highly fragmented and degraded. Once an organism dies, cellular processes such as DNA repair mechanisms are no longer active, and the unmaintained DNA degrades over time. Previous research has shown that ancient DNA is usually <100 base pairs (bp) long (e.g., Pääbo, 1989; Weyrich et al., 2017), and marine sedaDNA fragments also tend to be very short (~69 bp; Armbrecht et al., 2020). Although ancient DNA extracted from bones and teeth has been used extensively to study human and megafauna evolution for more than 30 years (Hagelberg et al., 2015), its application to marine settings is still relatively new. Consequently, sedaDNA laboratory protocols, as well as downstream bioinformatic processing and analysis of sedaDNA data, are not yet well established. This is especially the case for eukaryotes for which only trace amounts of sedaDNA are preserved in the forms of both extra- and intracellular DNA (e.g., within robust resting stages such as cysts and spores). Further, ancient DNA fragment size and damage analysis, standard procedures for validating ancient DNA signals in human- and megafauna-related ancient DNA research, have not yet been commonly applied to sedaDNA, making it difficult to evaluate sedaDNA authenticity across existing studies.
In contrast to ancient DNA, DNA from living organisms (modern DNA) is highly intact and overwhelmingly abundant in the environment, including the ocean. For example, the average size of the small subunit ribosomal RNA gene—SSU rRNA, a gene commonly used as a taxonomic marker—is approximately 20 times that of a typical ancient DNA molecule (~1,800 bp; Tanabe et al., 2016) and can occur in copy numbers of ~12,000 in a single cell of a marine phytoplankter (Zhu et al., 2005). Molecular biological techniques used to detect and investigate marine organisms in present-day ocean settings are well established and have greatly improved our knowledge about the functioning, composition, and dynamics of marine food webs (Amaral-Zettler et al., 2009; De Vargas et al., 2015; Carradec et al., 2018). In addition, metagenomic research of the modern ocean continues to generate invaluable reference sequences of living marine organisms to which ancient sequences can be compared.
Due to the sheer abundance of modern DNA in the ocean, contamination is a negligible issue in contemporary marine genomics because contaminant sequences are largely outweighed by the target genetic signal. The opposite pattern is the case for sedaDNA, however, and thorough contamination control is required at each step along the process of sedaDNA acquisition and analysis.
How Is sedaDNA Acquired from the Seafloor?
The recovery of sedaDNA from deep ocean sediments usually involves the acquisition of sediment cores from research vessels or platforms (Figure 1). Armbrecht et al. (2019) describe in detail such coring operations and best practice techniques. In brief, among the different coring methods currently available, gravity-based coring and advanced piston coring (APC) systems are preferred for sedaDNA analyses, as they recover the least disturbed sediments and minimize the likelihood of modern seawater ingress. Gravity-based corers simply “free fall” into the sediment, cutting a core from the seafloor. To trigger the APC system, hydraulic pressure is applied by pumping drill fluid toward the shear pins at the top of the core barrel (Sun et al., 2018). Only small volumes of drill fluid can enter the space between the core barrel and the collar from above after stroking, greatly reducing the risk of contamination. However, because the standard drill fluid is seawater, which inevitably contains vast amounts of modern marine organisms, contamination is still a risk, and strict contamination measures are necessary. This potential contamination source can be tracked by infusing a non-toxic, non-volatile chemical tracer (e.g., perfluoromethyldecalin, PFMD) at a constant rate into the drill fluid and testing sediment subsamples along the length of the core for the presence of the tracer (Figure 1).
When the core is on deck, measures can be taken to prevent contamination derived from core handling and the surrounding environment. Readily applied contamination control measures while working on board a research vessel or platform include wearing personal protective equipment; decontaminating the working environment, core liners, and cutting and splitting tools; instituting a variety of controls (e.g., air sampling, work bench swabbing, using PFMD controls alongside sedaDNA samples), and, if possible, working fast in cold, still-air conditions. Core storage under cold and anoxic conditions and/or freezing of sediment subsamples is also recommended, as well as subsequent sample processing at specialized ancient DNA facilities. The latter comprise ultraclean, low-DNA environments achieved by following a unidirectional workflow from sample preparation to extraction and sequencing library preparation in separate working areas, and positively pressured and frequently decontaminated laboratories (Fulton and Shapiro, 2019).
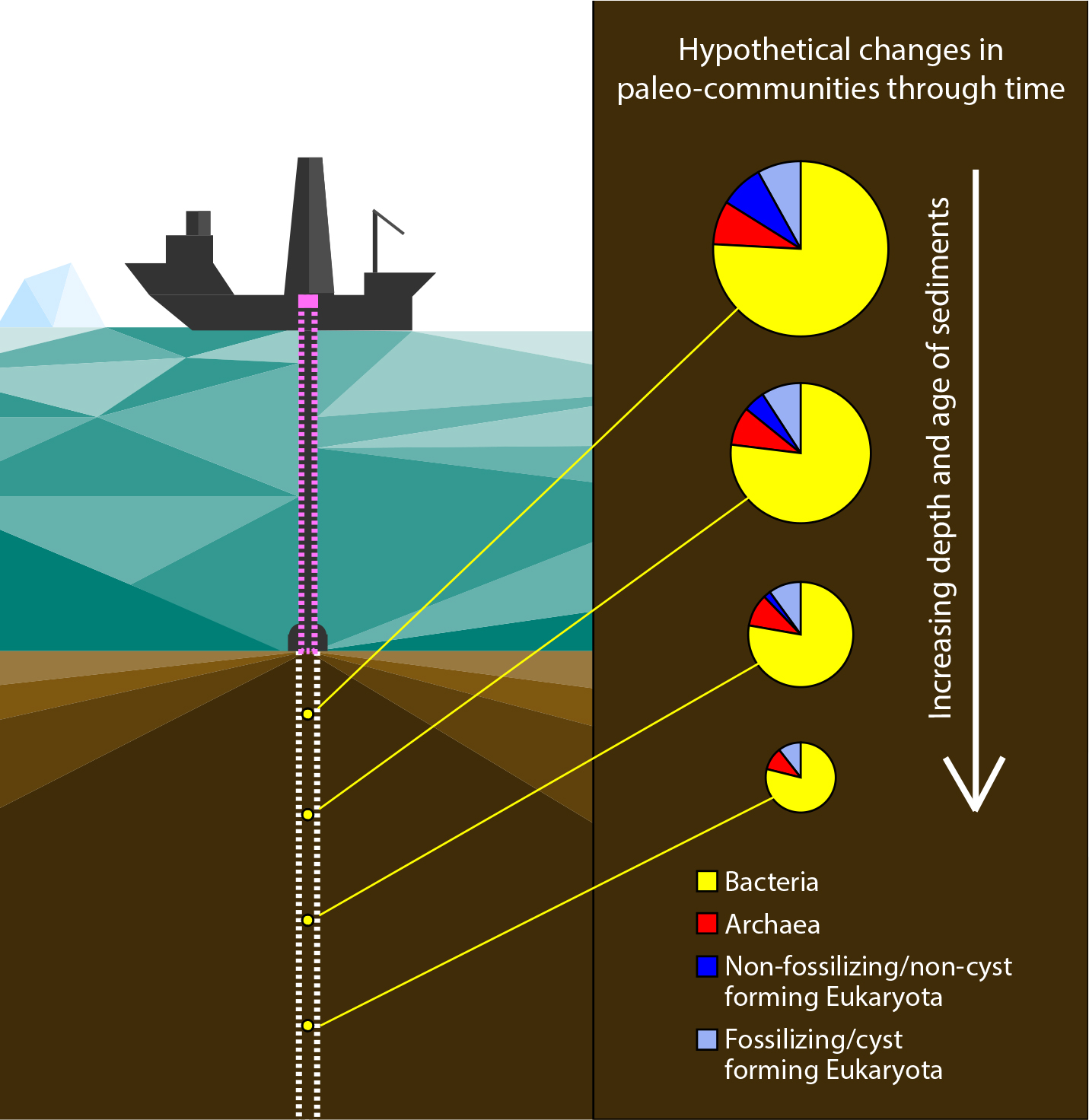
Figure 1. Schematic of a drilling vessel recovering a sediment core for sedaDNA analysis and hypothetical past marine community composition. The pink dashed line indicates the use of a chemical tracer for contamination tracking during coring. The white dashed line depicts the sediment core. Small yellow circles indicate theoretical sedaDNA sampling intervals, corresponding to pie charts on the right. Pie charts represent hypothetical paleo-communities detectable from sedaDNA shotgun analysis, where the majority (~75%, see text and Figure 3c) of the recovered sedaDNA sequences originate from bacteria, and where sedaDNA from fossilizing/cyst-forming taxa increases relative to non-fossilizing/non-cyst-forming taxa with subseafloor depth (assuming that sedaDNA of fossilizing/cyst-forming taxa preserves better than that of non-fossilizing/non-cyst-forming taxa). The decreasing size of the pie charts with subseafloor depth indicates an expected decrease in sedaDNA. Schematic not to scale. > High res figure
|
Are There Optimal Marine sedaDNA Extraction Techniques?
While a “one-fits-all” extraction protocol for sedaDNA would be highly desirable, optimized protocols are usually required, depending on the type of sediment and the target organisms to be analyzed. The efficiency of sedaDNA extraction can be highly variable, depending on sediment type and composition. Sediments rich in clay, borate, and organic content, and those kept at low temperatures and protected from oxygen and irradiation, usually contain relatively well preserved sedaDNA (Armbrecht et al., 2019, and references therein). A recent study showed that extracellular DNA binds well to clay minerals, which may protect it from being degraded by living bacteria in the subseafloor environment, and such DNA-substrate interactions are important to consider during sedaDNA extractions (Vuillemin et al., 2019).
Both intra- and extracellular sedaDNA can be extracted from sediments using a sequence of chemical and mechanical treatments. At the initial step of sedaDNA extractions, shaking the sample in a solution containing small beads (bead-beating) is commonly employed to break robust resting cells and to facilitate the recovery of intracellular as well as extracellular sedaDNA (e.g., Shaw et al., 2019). Phosphate-containing buffers have also been incorporated into sedaDNA extraction protocols, as phosphate has similar charge and structural properties to DNA so that it creates a competitive environment in which more DNA remains in solution, thereby aiding its isolation (Direito et al., 2012). We recently developed an optimized extraction protocol for marine eukaryote sedaDNA that combines two-step DNA-isolation (extracting DNA from fragile as well as robust organisms) with a DNA binding reaction in silica solution (Armbrecht et al., 2020). This technique facilitates recovery of particularly small and degraded DNA fragments (down to 27 bp), which is important for studies of ancient marine deposits, as sedaDNA fragmentation is expected to increase with the depth and age of the sediments.
Which Genomics and Sequencing Approaches Are Suitable for Studying sedaDNA?
Studies that rely on the analysis of sedaDNA should demonstrate data authenticity (i.e., the DNA recovered is ancient and free from modern contamination; Willerslev and Cooper, 2005). Validation protocols for ancient DNA include DNA damage analysis—for example, by applying mapDamage software, which was specifically developed to detect nucleotide misincorporations and fragmentation patterns that are characteristic of ancient DNA (Ginolhac et al., 2011; Jónsson et al., 2013). MapDamage has become a routine tool for authenticating ancient DNA across studies focusing on megafauna and humans (e.g., Llamas et al., 2015; Tobler et al., 2017). However, in highly complex metagenomic data, the identification and authentication of ancient sequences of very rare organisms remains challenging, partly due to the lack of high-quality reference sequences for the thousands of marine organisms thriving in the global ocean, and as the threshold of ~250 reads per species required to analyze and plot DNA damage patterns in mapDamage is often not reached (Collin et al., 2020). To overcome this issue, recent studies have focused on developing new bioinformatic techniques suitable for identification and authentication of low abundance ancient DNA in metagenomic data. For example, Hübler et al. (2019) developed HOPS (Heuristic Operations for Pathogen Screening) to screen for ancient pathogens in metagenomic samples, and Collin et al. (2020) described a new approach for processing and analyzing ancient metagenomic shotgun data focusing on the conservation of rare reads. The application and optimization of such tools for identifying and authenticating marine eukaryotes in sedaDNA appear highly promising and relevant to future sedaDNA research. A much less ideal but simpler authenticity assessment can be achieved through DNA fragment size analysis (as noted above, fragmentation is expected to increase with age of the sediment sample), which should be the minimum authenticity analysis undertaken in any marine sedaDNA study.
To date, however, most sedaDNA studies have used a metabarcoding approach to investigate paleocommunities. This commonly used method targets a specific DNA region used as a taxonomic marker to identify different species that are aggregated in a sediment sample (Taberlet et al., 2012). These genetic markers are amplified using primers (short sequences matching the start and end of the target gene) in a polymerase chain reaction (PCR) and are subsequently sequenced (Figure 2). This technique has been shown to be unsuitable for the study of ancient DNA (e.g., Weyrich et al., 2017) for the following reasons:
1. Ancient DNA is typically highly damaged so that primers may not bind.
2. The DNA segments to be amplified are usually longer (>100 bp) than most ancient DNA fragments (<100 bp; Pääbo, 1989), introducing biases toward longer sequences that favor modern contaminants where present, and skew the taxonomic composition.
3. PCRs are prone to inherent biases such as random amplification in the first few amplification cycles and PCR drift; hence, biases become more severe with increasing numbers of amplification cycles (Wagner et al., 1994; Taberlet et al., 2012) as necessitated with sedaDNA protocols.
4. The characteristic DNA damage patterns described above are no longer detectable in metabarcoding data as polymerases correct these patterns during amplification, preventing this mode of authenticity testing.
Studies that have applied metabarcoding should therefore be interpreted with caution unless they have shown authenticity of the sedaDNA through complementary analyses (e.g., fossils, biomarkers).
With ongoing increases in sequencing power and decreases in cost (Reuter et al., 2015), metagenomics is becoming a viable alternative to metabarcoding. Metagenomics studies extract and amplify the “total” DNA in a sample (i.e., potentially all species), thereby facilitating the recovery of DNA sequences proportionate to their original representation in that sample and independent of DNA fragment-size (“shotgun sequencing”; Figure 2). Thus, metagenomics approaches are better suited to studying sedaDNA, as they permit detection of bacteria, archaea, and eukaryotes, and they recover DNA damage patterns and fragment size variability without the biases inherent to metabarcoding. Community composition can then be reconstructed from this large pool of metagenomic data by screening for the occurrence of taxonomic marker genes. Additionally, metagenomic data sets offer the opportunity to draw functional information (“what organisms were doing”), as recently shown by Giosan et al. (2018), through their identification and use of chlorophyll biosynthesis proteins to estimate paleoproductivity. If the representation of the target organisms/genes is relatively low in the pool of total DNA data (e.g., in the case of eukaryote sedaDNA), very deep sequencing is required to recover sufficient genetic information to perform meaningful statistical analyses.
An attractive alternative that combines the specificity of the PCR approach but avoids the biases of the PCR method is the targeted enrichment of specific genetic sequences via hybridization-capture techniques (Horn, 2012). This approach uses short RNA probes (also called “baits,” analogous to the baits used in a fishing context) that are designed to be complementary to any DNA sequences the researchers may choose (e.g., specific genes of a target organism). By binding to the target sequence, these genetic baits “capture” DNA fragments in a manner that is akin to the PCR targeting, but independent of fragment size and with the preservation of damage patterns, allowing detailed authenticity testing (Figure 2). A recent study successfully applied this capture technique to Northern Hemisphere permafrost samples by developing PaleoChip Arctic 1.0 for investigating ancient Arctic plants and animals (Murchie et al., 2019).
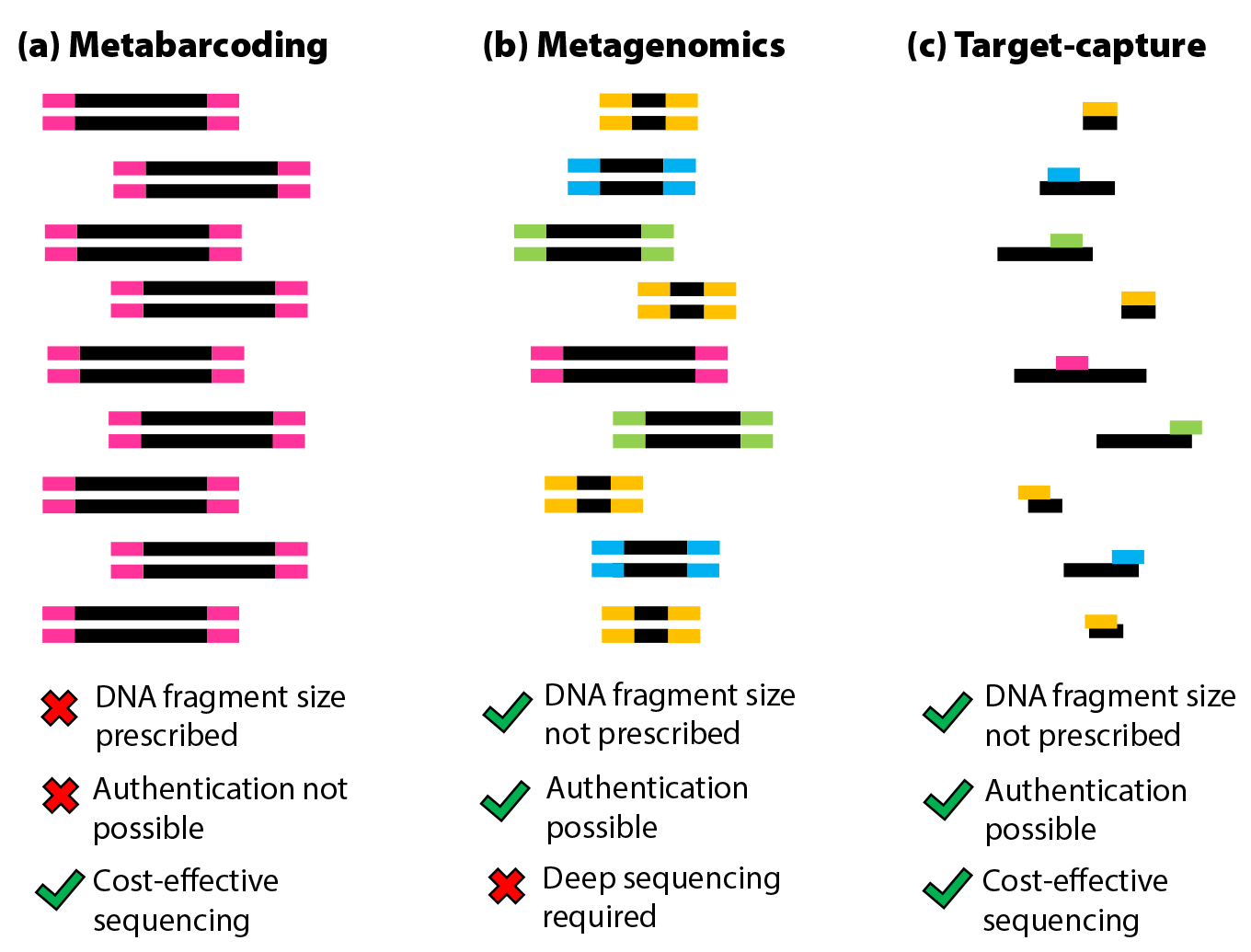
Figure 2. Schematic of different methodological approaches in modern and ancient marine genomics. (a) Metabarcoding is the amplification and analysis of equally sized DNA fragments from a total DNA extract. (b) Metagenomics is the extraction, amplification, and analysis of all DNA fragments independent of size. (c) Target-capture describes the enrichment and analysis of specific (chosen) DNA fragments independent of size from a total DNA extract. > High res figure
|
How Detailed Are the Community Data Generated from sedaDNA?
Most marine organisms are known genetically only by short segments of their genome, such as “taxonomic marker genes,” which occur in a large group of organisms but with slight variations in each species that allow taxonomic differentiation. An example are the ribosomal genes: small subunit ribosomal RNA (18S rRNA or SSU rRNA) or large subunit ribosomal RNA (28S rRNA or LSU rRNA). Applying metabarcoding of the 18S rRNA gene (V9 region only, ~130 bp long), De Vargas et al. (2015) estimated a taxa richness of ~150,000 eukaryotes in the modern global surface ocean, detecting over 3,000 diatoms alone (categorizing the latter as a “hyperdiverse” group). This estimate stands in comparison to only 12 complete diatom genomes currently available via the National Center for Biotechnology Information’s (NCBI) Genome database (https://www.ncbi.nlm.nih.gov/genome/; search terms: “Eukaryota”[Organism] AND “Bacillari-
ophyta”[Organism], June 16, 2020). The abundance of references for taxonomic marker genes over whole genomes of major marine eukaryotic taxa plainly illustrates why the use of marker genes for community composition assessments is popular.
Two of the best-known curated databases containing marine eukaryote sequences are the Protist Ribosomal Reference Database (PR2; Guillou et al., 2012) and the SILVA ribosomal RNA database (Quast et al., 2013). PR2 contains SSU rRNA sequences, while SILVA is split into two separate databases, one containing full-length sequences of the same gene (SSU rRNA) and the other full-length sequences of the LSU rRNA gene. Both SILVA databases contain sequence information from a variety of marine organisms—the latest release of the SILVA SSU database (SSURef NR 132) contains 592,561 bacterial, 25,026 archaeal, and 77,584 eukaryotic sequences, and the LSU database (LSURef 132) contains 168,075 bacterial, 1,440 archaeal, and 29,319 eukaryotic sequences (https://www.arb-silva.de/documentation/release-132/). Although PR2 and SILVA comprise extensive resources, it is important to consider the use of additional databases, depending on the target organisms and/or study focus. For example, in sedaDNA research, the NCBI database, the largest genetic database available (https://www.ncbi.nlm.nih.gov/), may also be considered as well as the Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP) database, an extensive resource for marine eukaryote RNA sequences (Keeling et al., 2014). While there is no standard approach to choosing a reference database (or combination of databases) for sedaDNA research, it is recommended to use one that is as complete as possible regarding reference sequences of the target organisms (Keeling et al., 2014; Collin et al., 2020).
Despite the usefulness of taxonomic marker genes for investigating marine eukaryotes, some limitations are yet to be overcome to achieve comprehensive community estimates and/or reconstructions. Even in modern marine investigations, part of the detected sequences can remain unidentified due to missing reference sequences, possibly because some organisms are (1) not easy or are impossible to culture and/or sequence, (2) rarely the subject of scientific investigations, or (3) entirely unknown and undescribed. For example, in their global study of marine eukaryotes, De Vargas et al. (2015) were able to assign taxonomy to two-thirds of the generated sequences, with the remaining third falling into the “unknown sequences” category. As a result of this imbalance in reference sequences across eukaryotes, some groups are better represented than others in genetic databases, which in turn will impact a study’s taxonomic resolution. Crucially, low taxonomic resolution can inhibit paleoenvironmental reconstruction efforts, where species-level identification is often pivotal in order to estimate key environmental or oceanographic conditions in a region of interest (Weckström et al., 2020).
An unfortunate consequence when applying taxonomic marker gene filters to metagenomic data is that the majority of the recovered sequencing data is not used. In fact, we recently showed that of a metagenomic marine sedaDNA shotgun data set, less than 1% of the quality-filtered sequencing reads were assigned taxonomy after alignment using the SILVA SSU reference database (Armbrecht et al., 2020). With high enough sequencing depth, a sufficient number of reads may remain available for statistical analyses; however, studies may wish to pursue optimizations in the experimental phase (e.g., hybridization-capture), or bioinformatic analysis (e.g., integrating complete databases), or both, where achieving high throughput is of concern.
Example of Increasing Taxonomic Resolution by Combining Information from Two Marker Genes
The following example shows how the rarely used LSU, when combined with the more widely used SSU, may provide increased taxonomic resolution in metagenomic sedaDNA data. Metagenomic sedaDNA sequence data, acquired from a sediment sample collected during the Sabrina Seafloor Survey 2017 (IN2017_V01) off East Antarctica (Armand et al., 2018), was extracted and analyzed using optimized laboratory and bioinformatic procedures described in detail in Armbrecht et al. (2020). The DNA fragment length distribution of the filtered sequences was analyzed and showed a predominance of very short sedaDNA fragments (76 bp), as expected (Figure 3a). The filtered sequences were aligned against both the SILVA SSURef NR 132 and the LSURef 132 databases (Huson et al., 2016). Read counts were determined for the SSU and LSU markers and analyzed separately, and then in combination, to avoid overrepresentation of any given taxon if detected by both. The read counts per taxon were summed if the taxon was identified exclusively by either SSU or LSU, and averaged if identified by both.
Using the combined markers showed a significant improvement in species resolution for eukaryotes (i.e., an increase in the number of eukaryotic taxa detected) relative to single markers (91 taxa determined using combined markers across all three samples vs. 59 and 51 for SSU and LSU alone, respectively; Figure 3b). Approximately 17% of the combined data was of eukaryote origin, including fossilizing as well as non-fossilizing taxa. For example, silica-skeleton-forming diatoms (Bacillariophyta) and Radiolaria were represented in the sedaDNA data, while non-fossilizing taxa such as cnidarians, copepods, and molluscs were also present (summarized under Eukaryota in Figure 3c). While the SSU appeared to be better suited for detecting major marine groups such as tintinnids (a group of ciliates), cnidarians, molluscs, and fish, the LSU provided better resolution for crustaceans (e.g., copepods) and haptophytes (e.g., Phaeophyceae). This example for a single sedaDNA sample shows that taxonomic resolution can be considerably improved when merging information from different taxonomic marker genes, and that the latter approach may be helpful for gaining a much more detailed understanding of marine paleocommunities in future sedaDNA studies.
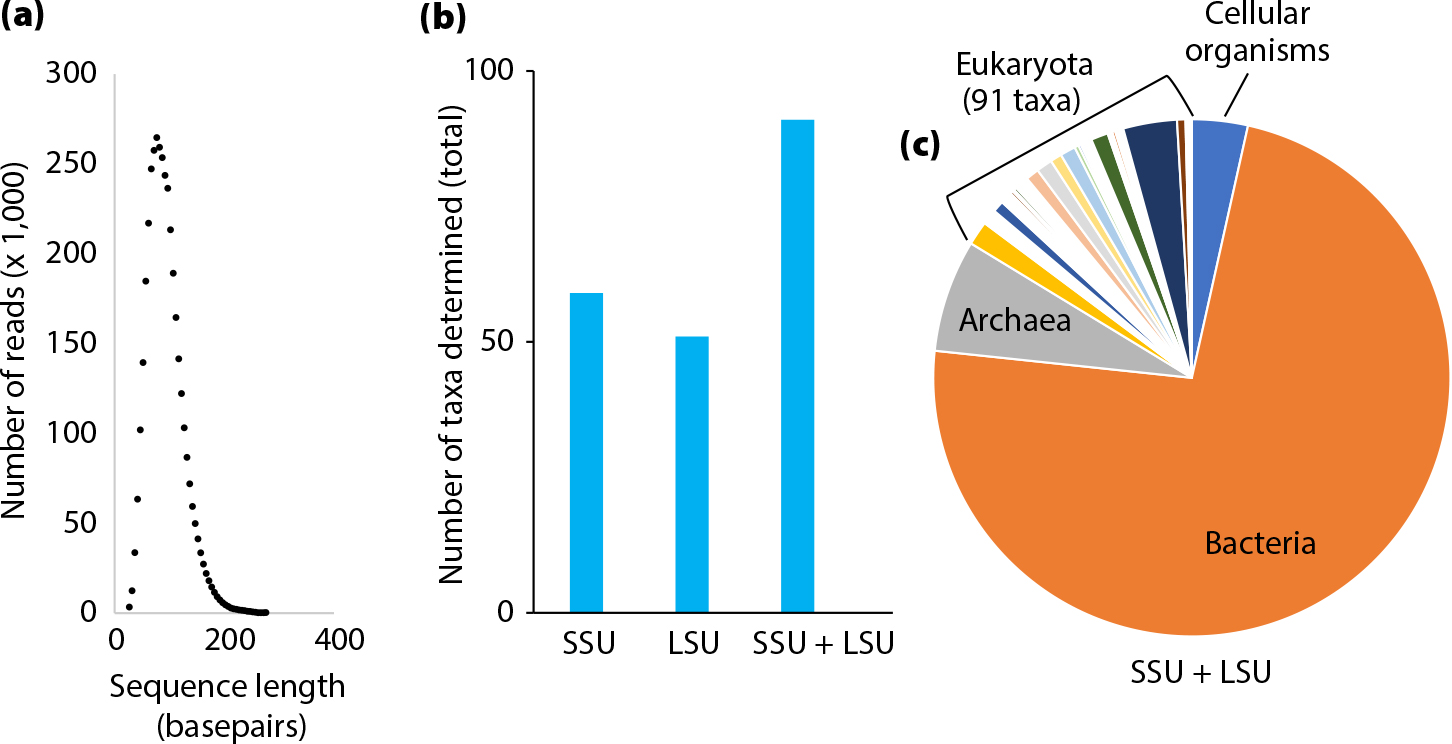
Figure 3. Taxonomic resolution achieved through different databases from short reads in metagenomic sedaDNA data. (a) Sequence-length distribution of a marine sedaDNA sample (counts of filtered reads classified into 51 DNA fragment size classes; sequenced on an Illumina HiSeq [2x 150 bp] at Garvan Institute for Medical Research, Sydney, Australia). (b) Total number of eukaryote taxa determined from metagenomic sedaDNA in a marine sediment sample using the SILVA small-subunit ribosomal RNA (SSU), large-subunit ribosomal RNA (LSU), and both SSU and LSU as reference databases. (c) Taxonomic composition derived from metagenomic sedaDNA of the same sample based on the combined SILVA SSU+LSU approach. > High res figure
|
What Does the Future Hold for Marine sedaDNA…
The emergence of the new marine sedaDNA research field has shown that genetic signals preserved in the marine seafloor are a precious reservoir of biological information that can be used for paleoceanographic reconstructions. However, several key points have been identified to consider in future sedaDNA research, outlined below.
…In Terms of Reference Sequences and Genetic Databases?
Obtaining a more detailed taxonomic picture of marine paleocommunities will rely closely upon the continuous addition of new reference sequences from modern marine organisms to taxonomic databases. This is achieved by both morphological and molecular characterization of individual species (e.g., through the application of single cell genomics), providing modern reference genomes against which ancient specimens can be aligned. Such references will be invaluable in efforts to achieve species-level community estimates and to detect indicator species of paleoenvironments using sedaDNA. Additionally, it is important to further optimize and develop bioinformatic tools that enable the streamlined analysis of the vast amounts of genetic data (millions of sequencing reads) generated from highly diverse marine sedaDNA samples. Currently, no standard bioinformatic approaches exist for the study of ancient marine sedaDNA, and their development should be a focus of future research to facilitate rapid analysis and comparison of results across studies and research groups.
…In Terms of Optimizations of Experimental Procedures?
sedaDNA extraction techniques may need further optimizations, depending on the study organisms. For example, diatoms are one of the most productive classes of phytoplankton in the Southern Ocean and are extremely useful paleoenvironmental indicators due to their sensitivity to changing oceanographic conditions and their excellent preservation as microfossils in marine sediments (Deppeler and Davidson, 2017). In contrast to their prevalence in the microfossil record, diatoms are often underrepresented in sedaDNA data (e.g., Shaw et al., 2019), which may be due to biases in DNA extraction methods, relatively poor preservation of ancient diatom DNA possibly linked to physical and chemical sediment characteristics, and/or current limitations in reference databases. Continuous improvement of extraction methods, application of targeted enrichment approaches, and addition of modern diatom sequences to genetic reference databases may help to tackle this important question in the future and enable detailed investigation of both diatoms and similar keystone marine species over geologic timescales.
…With Regard to Authenticity Testing of the Ancient Signal?
It will be important to move from metabarcoding to metagenomic techniques to allow authentication of ancient signals in genetic data (Taberlet et al., 2012; Weyrich et al., 2017; Collin et al., 2020). If the study organisms are expected to be a rare component of the metagenomic data, sequencing depth should be maximized, and sedaDNA damage-preserving target-enrichment techniques should be considered (Horn, 2012). Sequence-length distribution analyses offer a simple measure for assessing authenticity of sedaDNA data; however, DNA damage analysis should be applied in the future to test and ensure sedaDNA authenticity (Ginolhac et al., 2011; Jónsson et al., 2013; Huebler et al., 2019; Collin et al., 2020), especially when working with increasingly old sediment samples. Additionally, blank and environmental controls should be included in the processing and analysis of sedaDNA and lists of contaminant taxa made publicly available alongside each study’s results. This will allow interlaboratory comparisons of common contaminants introduced by reagents and/or databases, as well as laboratory or extraction method-specific contaminants, which might otherwise confound the results from which conclusions are drawn.
…For the Investigation of Species- and Location-Specific Degradation Patterns?
Preservation of marine sedaDNA in various locations should be investigated alongside environmental variables that might influence preservation and degradation patterns. Taxon-specific degradation patterns and taphonomic biases require in-depth investigation to determine whether calibrations of the community data are required in sedaDNA data. Much more research is required to determine the variations in sedaDNA fragment size by location, age, and taxonomic composition, and the environmental factors that contribute to sedaDNA degradation.
The points listed above are fundamental aspects to be addressed in future marine sedaDNA research, an area that is still in its infancy and that holds great potential for providing novel insights into the evolution and dynamics of marine ecosystems. Keeping these key points in mind is vital to continued efforts to reconstruct past marine communities from sedaDNA and will help to generate and authenticate better resolved data for exploring in detail the history of life in our ocean.
Acknowledgments
I thank Leanne Armand and Phil O’Brien, and the IN2017_V01 science party and crew (Commonwealth Scientific and Industrial Research Organisation [CSIRO]/Marine National Facility [MNF]) for their help in acquiring the sedaDNA sample that is exemplified in this article and for their input into this research. I also thank Gustaaf Hallegraeff, Chris Bolch, and Alan Cooper for supporting this work, as well as Holly Heiniger and Steve Johnson for technical assistance, and Raphael Eisenhofer, Kieren Mitchell, Laura Weyrich, Bastien Llamas, Ray Tobler, Vilma Pérez, Oscar Estrada-Santamaria, Yassine Souilmi, Yichen Liu, Steve Richards, João Teixeira, Christian Huber, Angad Johar, Joshua Schmidt, Salvador Herrando-Pérez, Jeremy Austin, Maria Lekis, and Adam Wilkins for many constructive discussions around sedaDNA. I am also grateful to Laurie Menviel and Katrin Meissner, the organizers of the International Conference on Paleoceanography 2019 (ICP13), where the idea for this manuscript was conceived. I was supported through the 2017 Australian Research Council Discovery Project DP170102261, by the University of Adelaide School of Biological Sciences, and by Past Global Changes (PAGES). Detailed information on eukaryote, bacterial, and archaeal communities not shown in this paper is available on request.

